forked from statOmics/PSLS
-
Notifications
You must be signed in to change notification settings - Fork 0
/
Copy path08-experimentalDesign3.Rmd
591 lines (425 loc) · 19.2 KB
/
08-experimentalDesign3.Rmd
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
238
239
240
241
242
243
244
245
246
247
248
249
250
251
252
253
254
255
256
257
258
259
260
261
262
263
264
265
266
267
268
269
270
271
272
273
274
275
276
277
278
279
280
281
282
283
284
285
286
287
288
289
290
291
292
293
294
295
296
297
298
299
300
301
302
303
304
305
306
307
308
309
310
311
312
313
314
315
316
317
318
319
320
321
322
323
324
325
326
327
328
329
330
331
332
333
334
335
336
337
338
339
340
341
342
343
344
345
346
347
348
349
350
351
352
353
354
355
356
357
358
359
360
361
362
363
364
365
366
367
368
369
370
371
372
373
374
375
376
377
378
379
380
381
382
383
384
385
386
387
388
389
390
391
392
393
394
395
396
397
398
399
400
401
402
403
404
405
406
407
408
409
410
411
412
413
414
415
416
417
418
419
420
421
422
423
424
425
426
427
428
429
430
431
432
433
434
435
436
437
438
439
440
441
442
443
444
445
446
447
448
449
450
451
452
453
454
455
456
457
458
459
460
461
462
463
464
465
466
467
468
469
470
471
472
473
474
475
476
477
478
479
480
481
482
483
484
485
486
487
488
489
490
491
492
493
494
495
496
497
498
499
500
501
502
503
504
505
506
507
508
509
510
511
512
513
514
515
516
517
518
519
520
521
522
523
524
525
526
527
528
529
530
531
532
533
534
535
536
537
538
539
540
541
542
543
544
545
546
547
548
549
550
551
552
553
554
555
556
557
558
559
560
561
562
563
564
565
566
567
568
569
570
571
572
573
574
575
576
577
578
579
580
581
582
583
584
585
586
587
588
589
590
591
---
title: "8.4 Experimental Design III: Replication and Power"
author: "Lieven Clement"
date: "statOmics, Ghent University (https://statomics.github.io)"
---
<a rel="license" href="https://creativecommons.org/licenses/by-nc-sa/4.0"><img alt="Creative Commons License" style="border-width:0" src="https://i.creativecommons.org/l/by-nc-sa/4.0/88x31.png" /></a>
```{r}
library(tidyverse)
```
# Concepts
- Experimental units representative for population: Randomisation
- Replication: technical vs biological, sample size - power
- Sources of variation: technical, biological, within subject, between subject
# Replication
Two pager on Replication in nature methods: [[PDF](https://www.nature.com/articles/nmeth.3091.pdf)]
{ width=100% }
## Example
Consider a single cell RNA-seq experiment where researchers want to assess the effect of drug treatment on gene expression
Potential research questions
- Is there a difference in average gene expression between treated and non treated samples
- Is there a difference in variability of gene expression between treated and non treated samples
## Sources of variation
TABLE 1 NATURE METHODS | VOL.11 NO.9 | SEPTEMBER 2014 | 879 - 880
| | Replicate type | Replicate category$^\text{a}$ |
|:---:|:---|:---:|
| | Colonies | B |
| | Strains | B |
| Animal study subjects | Cohoused groups | B |
| | Gender | B |
| | Individuals | B |
| | | |
| | Organs from sacrificed animals | B |
| | Methods for dissociating cells from tissue | T |
| Sample preparation | Dissociation runs from given tissue sample | T |
| | Individual cells | B |
| | | |
| | RNA-seq library construction | T |
| | Runs from the library of a given cell | T |
| Sequencing | Reads from different transcript molecules | V$^\text{b}$ |
| | Reads with unique molecular identifier (UMI) from a given transcript molecule | T |
(a) Replicates are categorized as biological (B), technical (T) or of variable type (V).
(b) Sequence reads serve diverse purposes depending on the application and how reads are used in analysis.
---
## At which level should we replicate?
- $\text{var}(X)=\sigma^2_A+\sigma^2_C+\sigma^2_M=\sigma^2_{TOT}$
- $\text{var}(\bar{X})=\frac{\sigma^2_A}{n_A}+\frac{\sigma^2_C}{n_A n_C} + \frac{\sigma^2_M}{n_A n_C n_M}$
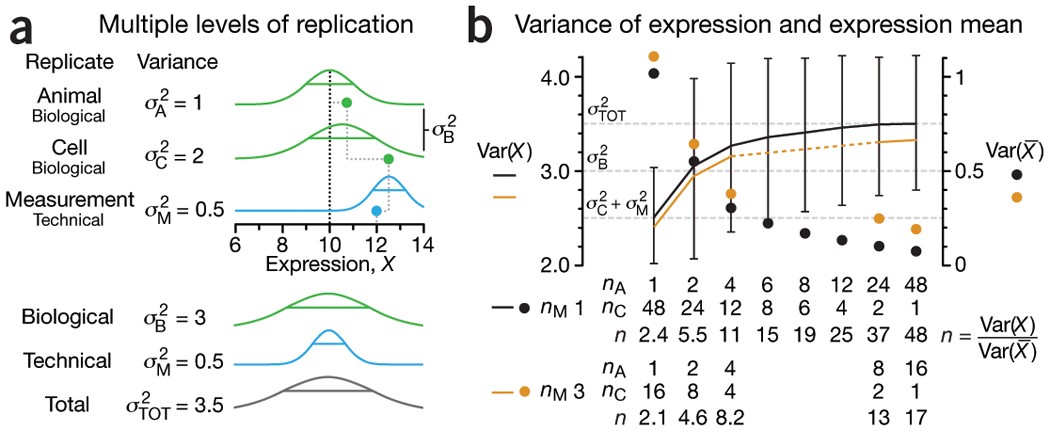
Figure 1 NATURE METHODS | VOL.11 NO.9 | SEPTEMBER 2014 | 879 - 880
(a) Three levels of replication (two biological, one technical) with animal, cell and measurement replicates normally distributed with a mean across animals of 10 and ratio of variances 1:2:0.5. Solid green (biological) and blue (technical) dots show how a measurement of the expression (X = 12) samples from all three sources of variation. Distribution s.d. is shown as horizontal lines.
(b) Expression variance, Var(X), and variance of expression mean, Var($\bar X$), computed across 10,000 simulations of nAnCnM = 48 measurements for unique combinations of the number of animals (nA = 1 to 48), cells per animal (nC = 1 to 48) and technical replicate measurements per cell (nM = 1 and 3). The ratio of Var(X) and Var($\bar X$) is the effective sample size, n, which corresponds to the equivalent number of statistically independent measurements. Horizontal dashed lines correspond to biological and total variation. Error bars on Var(X) show s.d. from the 10,000 simulated samples (nM = 1).
---
- Cost benefit trade-off: cost difference between biological and technical replicates
- Typically, biological variability is substantially larger than technical variability
$\rightarrow$ commit resources to sampling biologically relevant variables
$\rightarrow$ unless measures of technical variability are of interest then increasing number of technical measurements is valuable.
- Good experimental design practice includes planning for replication.
1. Identify the research questions to be answered with experiments.
2. Determine proportion of variability induced by each step.
3. Be aware for pseudoreplication and aim for statistically independent replicates.
---
# Power, sample size and other design aspects
Reading materials:
[Nature Methods (2013), 10(12), 1139–1140](https://www.nature.com/articles/nmeth.2738.pdf)
## Intermezzo linear regression in matrix form
- Linear regression is a very important statistical tool to study the association between variables and to build prediction models.
### Scalar form
- Consider a vector of predictors $\mathbf{x}=(x_1,\ldots,x_p)$ and
- a real-valued response $Y$
- then the linear regression model can be written as
\[
Y=f(\mathbf{x}) +\epsilon=\beta_0+\sum\limits_{j=1}^p x_j\beta_j + \epsilon
\]
with i.i.d. $\epsilon\sim N(0,\sigma^2)$
### Vector/Matrix form
- $n$ observations $(\mathbf{x}_1,y_1) \ldots (\mathbf{x}_n,y_n)$ with $\mathbf{x}_1^T=[1 x_1 \ldots x_p]$
- Regression in matrix notation
\[\mathbf{Y}=\mathbf{X\beta} + \mathbf{\epsilon}\]
with $\mathbf{Y}=\left[\begin{array}{c}y_1\\ \vdots\\y_n\end{array}\right]$,
$\mathbf{X}=\left[\begin{array}{cccc} 1&x_{11}&\ldots&x_{1p}\\
\vdots&\vdots&&\vdots\\
1&x_{n1}&\ldots&x_{np}
\end{array}\right]$ or $\mathbf{X}=\left[\begin{array}{c} \mathbf{x}_1^T\\\vdots\\\mathbf{x}_n^T\end{array}\right]$,
$\boldsymbol{\beta}=\left[\begin{array}{c}\beta_0\\ \vdots\\ \beta_p\end{array}\right]$ and
$\mathbf{\epsilon}=\left[\begin{array}{c} \epsilon_1 \\ \vdots \\ \epsilon_n\end{array}\right]$
---
## Least Squares (LS)
- Minimize the residual sum of squares
\begin{eqnarray*}
RSS(\boldsymbol{\beta})&=&\sum\limits_{i=1}^n e^2_i\\
&=&\sum\limits_{i=1}^n \left(y_i-\beta_0-\sum\limits_{j=1}^p x_{ij}\beta_j\right)^2
\end{eqnarray*}
- or in matrix notation
$$
\text{RSS}(\boldsymbol{\beta})=(\mathbf{Y}-\mathbf{X\beta})^T(\mathbf{Y}-\mathbf{X\beta})
$$
$$\rightarrow \hat{\boldsymbol{\beta}}=\text{argmin}_\beta \text{ RSS}(\boldsymbol{\beta})$$
---
### Minimize RSS
\[
\begin{array}{ccc}
\frac{\partial RSS}{\partial \boldsymbol{\beta}}&=&\mathbf{0}\\\\
\frac{(\mathbf{Y}-\mathbf{X\beta})^T(\mathbf{Y}-\mathbf{X}\boldsymbol{\beta})}{\partial \boldsymbol{\beta}}&=&\mathbf{0}\\\\
-2\mathbf{X}^T(\mathbf{Y}-\mathbf{X}\boldsymbol{\beta})&=&\mathbf{0}\\\\
\mathbf{X}^T\mathbf{X\beta}&=&\mathbf{X}^T\mathbf{Y}\\\\
\hat{\boldsymbol{\beta}}&=&(\mathbf{X}^T\mathbf{X})^{-1}\mathbf{X}^T\mathbf{Y}
\end{array}
\]
---
## Variance Estimator?
\[
\begin{array}{ccl}
\hat{\boldsymbol{\Sigma}}_{\hat{\boldsymbol{\beta}}}
&=&\text{var}\left[(\mathbf{X}^T\mathbf{X})^{-1}\mathbf{X}^T\mathbf{Y}\right]\\\\
&=&(\mathbf{X}^T\mathbf{X})^{-1}\mathbf{X}^T\text{var}\left[\mathbf{Y}\right]\mathbf{X}(\mathbf{X}^T\mathbf{X})^{-1}\\\\
&=&(\mathbf{X}^T\mathbf{X})^{-1}\mathbf{X}^T(\mathbf{I}\sigma^2)\mathbf{X}(\mathbf{X}^T\mathbf{X})^{-1}
\\\\
&=&(\mathbf{X}^T\mathbf{X})^{-1}\mathbf{X}^T\mathbf{I}\quad\mathbf{X}(\mathbf{X}^T\mathbf{X})^{-1}\sigma^2\\\\
%\hat{\boldmath{\Sigma}}_{\hat{\boldsymbol{\beta}}}&=&(\mathbf{X}^T\mathbf{X})^{-1}\mathbf{X}^T \text{var}\left[\mathbf{Y}\right](\mathbf{X}^T\mathbf{X})^{-1}\mathbf{X}\\
&=&(\mathbf{X}^T\mathbf{X})^{-1}\mathbf{X}^T\mathbf{X}(\mathbf{X}^T\mathbf{X})^{-1}\sigma^2\\\\
&=&(\mathbf{X}^T\mathbf{X})^{-1}\sigma^2
\end{array}
\]
The uncertainty on the model parameters thus depends on the residual variability and the design!
- The larger $\mathbf{X}^T\mathbf{X}$ the more information the experiment contains on the model parameters and the smaller their variances and standard errors will be!
- Factorial designs?
- Designs with continuous predictors?
---
The effect size of interest is typically a linear combinations of the model parameters, i.e.
$$
l_0 \times \beta_0 + l_1 \times \beta_1 + ... + l_{p-1} \times \beta_{p-1} = \mathbf{L}^T\boldsymbol{\beta}
$$
The null hypothesis of our test statistics is than formulated as
$$
H_0: \mathbf{L}^T\boldsymbol{\beta} = 0
$$
vs the alternative hypothesis
$$
H_0: \mathbf{L}^T\boldsymbol{\beta} \neq 0
$$
which can be assessed using a t-test statistic:
$$
t=\frac{\mathbf{L}^T\hat{\boldsymbol{\beta}} - 0}{\text{se}_{\mathbf{L}^T\hat{\boldsymbol{\beta}}}}
$$
which follows a t-distribution with n-p degrees of freedom under the null hypothesis when all assumptions for the linear model are valid.
So the power
$$P(p < 0.05 | H_1)$$
will typically depends on
- the real effect size in the population $\mathbf{L}^T\boldsymbol{\beta}$.
- Number of observations: SE and df of t-test.
- Choice of the design points.
- Choice of significance level.
Similar to the introductory example, we can use simulations to assess the power.
## Mouse example
- In 2021 Choa et al. published that the cytokine Thymic stromal lymphopoietin
(TSLP) induced fat loss through sebum secretion (talg). [[html](https://www.science.org/doi/full/10.1126/science.abd2893)] [[PDF](https://www.science.org/doi/pdf/10.1126/science.abd2893)]
- Suppose that you would like to set up a similar study to test if cytokine interleukin 25 (IL) also has beneficial effect.
- You plan to setup a study with a control group of high fat diet (HFD) fed mice and a treatment group that recieves the HFD and IL.
- What sample size do you need to pick up the effect of the treatment.
### How will we analyse the data of this experiment?
---
- $H_0$: The average weight difference is equal to zero
- $H_1$: The average weight difference is different from zero
- Two sample t-test or a t-test on the slope of a linear model with one dummy variable.
$$ Y_i = \beta_0 + \beta_1 X_\text{iIL} + \epsilon_i$$
$$
\text{with }
X_\text{iIL}=\begin{cases}X_{iIL}=0 & \text{HFD}\\X_{iIL}=1 & \text{HFD + IL}\end{cases}.
$$
- Estimated effect size
$$\hat\delta = \bar X_{IL} - \bar X_{c} = \hat \beta_1$$
- Test statistic
$$
T = \frac{\bar{X}_{IL}-\bar{X}_{c}}{SD_\text{pooled} \times \sqrt{\frac{1}{n_1} + \frac{1}{n_2}}} = \frac{\hat\beta_1}{\text{SE}_{\hat\beta_1}}
$$
- $\hat \beta_1$ is an unbiased estimator of the real weight difference ($\delta$) that would occur in the population of rats fed with HFD and rats fed with HFD + IL.
### Power?
$$
P[p < \alpha \vert \beta_1 \neq 0]
$$
- Hence, the power will depend on the real weight difference between the group means $\beta_1$ in the population.
- The variability of the weight measurements
- The significance level $\alpha$
- Sample size $n_{IL}$ and $n_c$ in both groups
We can estimate the power if
1. The assumptions of the model are met: weights are normally distributed with same variance
and we know
2. the standard deviation of the weight measurements around their average mean for HFD-fed mice
3. the real effect size in the population
4. $n_1$ and $n_2$
### Use data from a previous experiment to get insight in mice data
Suppose that we have access to the data of a preliminary experiment (e.g. provided by Karen Svenson via Gary Churchill and Dan Gatti and partially funded by P50 GM070683 on [PH525x](http://genomicsclass.github.io/book/pages/random_variables.html))
```{r}
mice <- read.csv("https://raw.githubusercontent.com/genomicsclass/dagdata/master/inst/extdata/femaleMiceWeights.csv")
mice %>%
ggplot(aes(x = Diet, y = Bodyweight)) +
geom_boxplot(outlier.shape = FALSE) +
geom_jitter()
mice %>%
ggplot(aes(sample = Bodyweight)) +
geom_qq() +
geom_qq_line() +
facet_wrap(~Diet)
```
```{r}
mice <- mice %>% mutate(Diet = as.factor(Diet))
miceSum <- mice %>%
group_by(Diet) %>%
summarize(
mean = mean(Bodyweight, na.rm = TRUE),
sd = sd(Bodyweight, na.rm = TRUE),
n = n()
) %>%
mutate(se = sd / sqrt(n))
miceSum
```
In the experiment we have data from two diets:
- regular diet of cerial and grain based diet (Chow)
- High Fat (hf)
We can use the hf mice as input for our power analysis.
- The data of the hf mice seem to be normally distributed
- The mean weight is `r miceSum %>% filter(Diet == "hf") %>% pull("mean") %>% round(.,1)`g
- The SD of the weight is `r miceSum %>% filter(Diet == "hf") %>% pull("sd") %>% round(.,1)`g
Effect size?
- The alternative hypothesis is complex.
- It includes all possible effects!
- In order to do the power analysis we will have to choose a minimum effect size that we would like to detect.
- Suppose that we would like to detect if the weight of the rats difference $\delta$ of at least 10%.
```{r}
delta <- -round(miceSum$mean[2] * .1, 1)
delta
```
Note, that the average weight then would get close to that of the rats in our pilot experiment that were fed on the chow diet.
We can set up a simulation study to assess the power of an experiment with 8 mice in each group:
#### One simulation
```{r}
set.seed(1423)
n1 <- n2 <- 8
b0 <- round(miceSum$mean[2], 1)
b1 <- -delta
sd <- round(miceSum$sd[2], 1)
alpha <- 0.05
x <- rep(0:1, c(n1, n2))
y <- b0 + b1 * x + rnorm(n1 + n2, 0, sd = sd)
fit <- lm(y ~ x)
bhat <- fit$coef
stat <- summary(fit)
summary(fit)$coef[2, ]
```
For this simulated experiment we would `r ifelse(summary(fit)$coef[2,"Pr(>|t|)"] < alpha,"","not")` have been able to pick up the effect of the treatment at the significance level $\alpha$=`r alpha` (p = `r round(summary(fit)$coef[2,"Pr(>|t|)"],2)`)!
- We have to repeat the experiment many times to estimate the power.
- We will therefore put our simulation in a general function.
#### Simulation of repeated experiments
```{r}
library(multcomp)
n1 <- n2 <- 8
b0 <- round(miceSum$mean[2], 1)
b1 <- -delta
sd <- round(miceSum$sd[2], 1)
predictorData <- data.frame(Diet = rep(c("c", "hf"), c(n1, n2)) %>% as.factor())
alpha <- 0.05
simLm <- function(form, data, betas, sd, contrasts, simIndex = NA) {
dataSim <- data
X <- model.matrix(form, dataSim)
dataSim$ySim <- X %*% betas + rnorm(nrow(dataSim), 0, sd)
form <- formula(paste("ySim ~", form[[2]]))
fitSim <- lm(form, dataSim)
mcp <- glht(fitSim, linfct = contrasts)
return(summary(mcp)$test[c("coefficients", "sigma", "tstat", "pvalues")] %>% unlist())
}
simLm(
form = ~Diet,
data = predictorData,
betas = c(b0, b1),
sd = sd,
contrasts = "Diethf = 0"
)
```
We now have a generic function that can simulate data from a normal distribution for every design.
The function has arguments:
- `form`: One sided formula including the structure of the predictors in the model
- `data`: Data frame with the predictor values for the design
- `betas`: A vector with values for all mean model parameters
- `sd`: The standard deviation of the error
- `contrasts`: a scalar or a vector with the null hypotheses that we would like to assess.
- `simIndex`: an arbitrary argument that is not used by the function but that will allow it to be used in an sapply loop that runs from 1 up to the number of simulations.
Simulate nSim = 1000 repeated experiments:
```{r}
set.seed(1425)
nSim <- 1000
simResults <- t(sapply(1:nSim, simLm, form = ~Diet, data = predictorData, betas = c(b0, b1), sd = sd, contrasts = "Diethf = 0"))
power <- mean(simResults[, 4] < alpha)
power
```
We have a power of `r power*100`% to pick up the treatment effect when using 8 bioreps in each treatment group.
#### Power for multiple sample sizes
We can now calculate the power for multiple sample sizes.
```{r}
power <- data.frame(n = c(3, 5, 10, 25, 50, 75, 100), power = NA)
for (i in 1:nrow(power))
{
n1 <- n2 <- power$n[i]
predictorData <- data.frame(Diet = rep(c("c", "hf"), c(n1, n2)) %>% as.factor())
simResults <- t(sapply(1:nSim, simLm, form = ~Diet, data = predictorData, betas = c(b0, b1), sd = sd, contrasts = "Diethf = 0"))
power$power[i] <- mean(simResults[, "pvalues"] < alpha)
}
power
```
```{r}
power %>%
ggplot(aes(x = n, y = power)) +
geom_line()
```
#### Assess power in function of sample size for different effect sizes
Note, that
- the sign of the delta is arbitrary because we test two-sided.
- the intercept is arbitrary because we only asses $\beta_0$
- We therefore typically set $\beta_0 = 0$
```{r}
nSim <- 1000
b0 <- 0
deltas <- c(1, 2, 3, 5, 10)
sd <- round(miceSum$sd[2], 1)
ns <- c(3, 5, 10, 20, 25, 50, 75, 100)
power <- data.frame(
b1 = rep(deltas, each = length(ns)),
n = rep(ns, length(deltas)),
power = NA
)
for (i in 1:nrow(power))
{
b1 <- power$b1[i]
n1 <- n2 <- power$n[i]
predictorData <- data.frame(Diet = rep(c("c", "hf"), c(n1, n2)) %>% as.factor())
simResults <- t(sapply(1:nSim, simLm, form = ~Diet, data = predictorData, betas = c(b0, b1), sd = sd, contrasts = "Diethf = 0"))
power$power[i] <- mean(simResults[, "pvalues"] < alpha)
}
```
```{r}
power %>%
ggplot(aes(x = n, y = power, col = b1 %>% as.factor())) +
geom_line()
```
Note, that the power curves are still a bit choppy because we selected a limited number of sample sizes and because `nSim` simulations is not enough to get a good power estimate when the power is low.
# Final remarks
- The code can be easily extended towards other designs by altering
- predictor data
- formula
- contrast
- The simulations start to take long when you evaluate many scenario's.
- More efficient code using matrices
- For a two group comparison closed form solutions exist
## More efficient code based on matrix algebra
Code runs much faster. We now simulate 20 times more experiments in a much shorter time!
```{r}
simFast <- function(form, data, betas, sd, contrasts, alpha = .05, nSim = 10000) {
ySim <- rnorm(nrow(data) * nSim, sd = sd)
dim(ySim) <- c(nrow(data), nSim)
design <- model.matrix(form, data)
ySim <- ySim + c(design %*% betas)
ySim <- t(ySim)
### Fitting
fitAll <- limma::lmFit(ySim, design)
### Inference
varUnscaled <- c(t(contrasts) %*% fitAll$cov.coefficients %*% contrasts)
contrasts <- fitAll$coefficients %*% contrasts
seContrasts <- varUnscaled^.5 * fitAll$sigma
tstats <- contrasts / seContrasts
pvals <- pt(abs(tstats), fitAll$df.residual, lower.tail = FALSE) * 2
return(mean(pvals < alpha))
}
```
```{r}
nSim <- 20000
b0 <- 0
sd <- round(miceSum$sd[2], 1)
ns <- c(3, 5, 10, 20, 25, 50, 75, 100)
deltas <- c(1, 2, 3, 5, 10)
contrast <- limma::makeContrasts("Diethf", levels = c("(Intercept)", "Diethf"))
powerFast <- matrix(NA, nrow = length(ns) * length(deltas), ncol = 3) %>% as.data.frame()
names(powerFast) <- c("b1", "n", "power")
form <- ~Diet
i <- 0
for (n in ns)
{
n1 <- n2 <- n
### Simulation
predictorData <- data.frame(Diet = rep(c("c", "hf"), c(n1, n2)) %>% as.factor())
for (b1 in deltas)
{
i <- i + 1
betas <- c(b0, b1)
powerFast[i, ] <- c(b1, n, simFast(form, predictorData, betas, sd, contrasts = contrast, alpha = alpha, nSim = nSim))
}
}
```
```{r}
powerFast %>%
ggplot(aes(x = n, y = power, col = b1 %>% as.factor())) +
geom_line()
```
## More efficient code based on closed form solution that exists for two group comparison
For the two sample t-test a closed form estimate exists for the power.
In this context the Cohen's effect size is typically used:
$D = \frac{\delta}{SD}$
```{r}
power.t.test(n = 8, delta = delta, sd = sd, type = "two.sample")
```
Note, that this is very similar to the power that we calculated using the simulations!
```{r}
b0 <- 0
sd <- round(miceSum$sd[2], 1)
ns <- c(3, 5, 10, 20, 25, 50, 75, 100)
deltas <- c(1, 2, 3, 5, 10)
powerTheo <- data.frame(
deltas = rep(deltas, each = length(ns)),
n = rep(ns, length(deltas)),
power = NA
)
powerTheo$power <- apply(powerTheo[, 1:2], 1, function(x) power.t.test(delta = x[1], n = x[2], sd = sd, type = "two.sample")$power)
```
```{r}
powerTheo %>%
ggplot(aes(x = n, y = power, col = deltas %>% as.factor())) +
geom_line()
```